| Академия » Статьи » Лекции » Анатомия |
СТРОМАЛЬНО-СОСУДИСТЫЕ ДИСТРОФИИ
|
СТРОМАЛЬНО-СОСУДИСТЫЕ ДИСТРОФИИ: МОРФОЛОГИЯ СИСТЕМНОЙ ПРОГРЕССИРУЮЩЕЙ ДЕЗОРГАНИЗАЦИИ СОЕДИНИТЕЛЬНОЙ ТКАНИ Стромально-сосудистые (мезенхимальные) дистрофии развиваются в результате нарушений обмена в соединительной ткани и выявляются в строме органов и стенках сосудов. Стромально-сосудистые дистрофии развиваются на территории гистиона, который, как известно, (образован отрезком микроциркуляторного русла с окружающими его элементами соединительной ткани (основное вещество, волокнистые структуры, клетки) и нервными волокнами. Понятным становится в связи с этим преобладание среди механизмов развития стромально-сосудистых дистрофий нарушений транспортных систем трофики, общности морфогенеза, возможности не только сочетания различных видов дистрофий, но и перехода одного вида в другой. Эти особенности стромально-сосудистых дистрофий ярко выражены при диспротеин0зах. Они-то и составляют сущность системной прогрессирующей дезорганизации соединительной ткани. В одних случаях в основе этой дезорганизации лежит прогрессирующая деструкция соединительной ткани — прогрессирующая дезорганизация как следствие деструкции соединительной ткани, в других — синтез аномального белка — прогрессирующая дезорганизация как следствие синтеза аномального белка в соединительной ткани. СИСТЕМНАЯ ПРОГРЕССИРУЮЩАЯ ДЕЗОРГАНИЗАЦИЯ СОЕДИНИТЕЛЬНОЙ ТКАНИ КАК СЛЕДСТВИЕ ЕЕ ДЕСТРУКЦИИ Этот вид системной дезорганизации обусловлен в большинстве случаев инфекцией, чаще стрептококковой, и тяжелыми иммунными (аутоиммунными) нарушениями, которые наиболее ярко выражены при ревматических заболеваниях./Определенное значение в их возникновении имеют и наследственные факторы. Начальные изменения системной дезорганизации соединительной ткани находят в парапластическвй субстанции (основное вещество соединительной ткани), где накапливаются гликозаминогликаны (хромотропные вещества), главным образом за счет гидрофильных гиалуроновых структур, а также плазменные белки, преимущественно глобулины. Накопление гликозаминогликанов связано с активной деятельностью фибробластов, а глобулинов — как с усиливающейся гидрофильностью соединительной ткани, так и с нарастающей плазморрагией. Коллагеновые фибриллы практически не страдают, если не считать некоторого их разволокнения; однако "упаковка" коллагеновых фибрилл в волокне становится более рыхлой. В 1961 г. А.И.Струков назвал эти изменения мукоидным набуханием, поверхностной фазой дезорганизации соединительной ткани. По существу он дал новое толкование миксоматозного (хромотропного) отека соединительной ткани, описанного В.Т.Талалаевым при ревматизме (1923). Однако термин "мукоидное набухание" за рубежом не прижился; обычно говорят о талалаевском мукоидном или хромотропном отеке. При мукоидном набухании часто развиваются клеточные реакции в виде лимфоцитарно-макрофагальных скоплений (полиморфно-ядерные лейкоциты редки), что свидетельствует об участии иммунных реакций при этой поверхностной и обратимой дезорганизации соединительной ткани. Если учесть, что при ревматических Заболеваниях (особенно ревматизме и ревматоидном артрите) иммунные реакции индуцированы стрептококком, с чем связано резкое повышение активности гиалуронидаз, то накопление гиалуроновых структур в соединительной ткани в этой ситуации можно рассматривать как проявление адаптации. Несмотря на общие пато- и морфогенетические механизмы мукоидного набухания при ревматических заболеваниях, оно имеет определенное своеобразие в зависимости от особенностей этиологии и патогенеза каждого из заболеваний; следует учитывать также и структурно-функциональные особенности органов, что показано при ревматизме, системной красной волчанке, ревматоидном артрите. Поверхностная дезорганизация соединительной ткани обычно прогрессирует, становится глубокой, захватывающей как интерстициальное (межуточное) вещество, так и коллагеновые структуры — развиваются фибриноидные изменения, или фибриноидное набухание. Это уже необратимый процесс, завершающийся фибриноидным некрозом, гиалинозом, склерозом. E.Neumann, впервые описавший фибриноидное набухание (1896) считал, что в его основе лежат повреждение коллагеновых волокон и приобретение ими свойств фибрина. Так появилось понятие о фибриноиде — веществе, которое возникает при фибриноидном набухании и отличается по многим свойствам от коллагена и фибрина. В дальнейшем было показано, что в формировании фибриноида, помимо деструкции коллагеновых волокон, большую роль играет состояние основного вещества, прежде всего его гликозаминогликанов, которые способны осаждаться щелочными белками, высвобождающимися при повреждении волокнистых и клеточных структур соединительной ткани. Было установлено, что в построении фибриноида принимают участие и белки плазмы, в первую очередь фибриноген с последующим превращением его в фибрин. Деление фибриноида на два вида — с фибрином и без фибрина, было опровергнуто результатами как биохимических, иммуногистохимических, рак и поляризационно-микроскопических и электронно-микроскопических исследований. Фибрин — обязательный компонент фибриноида, что лишний раз свидетельствует о важной роли плазморрагии в его развитии. Обнаружение же в фибриноиде иммунных комплексов подтверждает важную роль не только плазморрагии, но и нарушений иммунологического гомеостаза при фибриноидных изменениях. Становится понятной клеточная реакция при фибриноидных изменениях, преимущественно плазмоцитарная и лимфоцитарно-макрофагальная (в случаях фибриноидного некроза и нейтрофильная). В ответ на деструкцию возникает воспаление по сути своей иммунное [Струков А.И., 1979]. При нарушениях иммунологического гомеостаза образование фибриноида обусловлено главным образом иммунокомплексным повреждением соединительной ткани с последующей абсорбцией фибрина — "фибриноид иммунных комплексов", "фибриноид деструкции". Однако при различных ревматических болезнях, патогенез которых связан с нарушением иммунологического гомеостаза, образуется разный фибриноид. Так, анализ фибриноидных изменений сердца при ревматизме показал, что расщепление коллагеновых фибрилл, как и изменения склеивающего вещества коллагеновых волокон, может быть причиной последующей инсудации плазменных белков, прежде ^сего фибриногена, при этом поврежденные коллагеновые волокна приобретают тинкториальные свойства фибрина. В очагах фибриноида обнаруживают фибрин, иммуноглобулины, комплемент. Подобную характеристику имеет и фибриноид при ревматоидном артрите. При системной красной волчанке — классической иммунокомплексной болезни — фибриноид имеет свою особенность. В фибриноиде определяется ядерный материал, что связано со своеобразием иммунных комплексов — высокая комплементсвязывающая активность комплексов, их лизирующие, ленкотаксические и хемотаксические свойства, а главное, антинуклеарная направленность комплексов. Системный гиалиноз как сосудов, особенно микроциркуляции, так и собственно соединительной ткани завершает процессы системной ее дезорганизации. Сложный гиалин, среди микрофибрилл которого видны разрушенные элементы соединительной ткани, — это "гиалин деструкции", так как основой его является обычно "фибриноид". Поэтому сложный гиалин отличает высокое содержание фибрина, наличие в нем иммунных комплексов. Сложный галин и "старый" фибриноид имеют много общих черт. Это понятно, так как к их образованию причастны одни и те же иммунопатологические механизмы. ■ Итак, системная дезорганизация соединительной ткани как следствие ее прогрессирующей деструкции представлена следующими последовательно сменяющими друг друга процессами, составляющими сущность стромально-сосудистых (мезенхимальных) диспротеинозов: ▲ мукоидным набуханием (нарушение обмена белков и гликозаминогликанов основного вещества, плазморрагия, набухание коллагеновых волокон); ▲ фибриноидным набуханием с образованием фибриноида (деструкция коллагеновых волокон и основного вещества, плазморрагия, образование белково-полисахаридных комплексов); ▲ гиалинозом (деструкция коллагеновых волокон и основного вещества, плазморрагия, преципитация белков плазмы, образование белка гиалина) (схема 4). Схема 4. Морфогенез системной прогрессирующей дезорганизации соединительной ткани вследствие ее деструкции 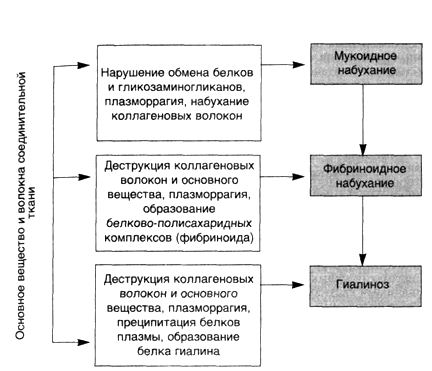 На основании приведенных материалов можно убедиться в том, что ревматические болезни отличаются от других системных заболеваний прежде всего двумя признаками — особенностью патогенеза и своеобразием морфогенеза. Особенность патогенеза заключается в ведущей роли при этих заболеваниях иммунопатологических механизмов. Морфогенетическое своеобразие заключается в системной дезорганизации соединительной ткани в результате прогрессирующей ее деструкции. При этом выявляется одна особенность — преимущественное поражение при том или ином ревматическом заболевании определенных, характерных только для этого заболевания, органов. При ревматизме такими органами-мишенями становятся сердце и сосуды, при ревматоидном артрите — суставы, при системной красной волчанке — микроциркуляторное русло и почки, при склеродермии (прогрессирующий системный склероз) — кожа и соединительная ткань ряда органов, при узелковом периартериите — артерии, пои дерматомиозите — кожа и мышцы. Эти шесть болезней, дополненные в последнее время болезнью Шегрена, образуют группу ревматических (бывших коллагеновых) заболеваний. Эта группа, как видно, четко очерчена патогенезом и морфогенезом. К сожалению, в последние годы морфогенез — основа патогенеза ревматических болезней — был перечеркнут, что привело к "разрастанию" и безликости группы ревматических болезней. Отрицание морфогенетических (морфологических) признаков заболеваний перечеркивает и признанный общей патологией постулат Н.Н.Аничкова : "Морфогенез — основа патогенеза". Отказ от этого принципа ведет к необоснованным заключениям, которые, как говорили великие энциклопедисты прошлого, хуже, чем заблуждение. СИСТЕМНАЯ ПРОГРЕССИРУЮЩАЯ ДЕЗОРГАНИЗАЦИЯ СОЕДИНИТЕЛЬНОЙ ТКАНИ КАК СЛЕДСТВИЕ СИНТЕЗА АНОМАЛЬНОГО БЕЛКА Этот вид системной прогрессирующей дезорганизации соединительной ткани представлен своеобразным стромально-сосудистым (мезенхимальным) диспротеинозом — системным, или генерализованным, амилоидозом. В 1844 г. венский патолог К.Рокитанский описал изменения паренхиматозных органов, которые, становясь плотными и ломкими, приобретали сальный вид. Так возникло понятие о "сальной болезни" Рокитанского. Несколько лет спустя Р.Вирхов назвал вещество, появляющееся в органах при "сальной болезни", амилоидом, поскольку под действием йода и серной кислоты оно, подобно крахмалу, окрашивалось в синий цвет. Потребовалось 20 лет для того, чтобы опровергнуть утверждения Р.Вирхова и доказать белковую природу амилоида [Руднев М.М. и Klihne W., 1865]. Амилоид — гликопротеид, основным компонентом которого является фибриллярный белок (F-компонент), имеющий характерную ультраструктуру. F-компонент связан в амилоиде с плазменными глюкопротеидами (Р-компонент). Оба эти компонента обладают антигенными свойствами. Амилоидное вещество метахроматично, дает характерную люминесценцию с тиофлавинами. Белок амилоидных фибрилл синтезируется клетками, которые получили название "амилоидобласты" (В.В.Серов). Они представлены при генерализованном амилоидозе макрофагами, плазматическими клетками, кардиомиоцитами, гладкими мышечными клетками сосудов и др. Фибриллы амилоида образуются на цитолемме амилоидобластов в ее инвагинатах. Образующееся амилоидное вещество выпадает по ходу ретикулярных (периретикулярный амилоидоз) либо коллагеновых (периколлагенивый амилоидоз) волокон. Встречается и смешанный вариант выпадения амилоида. Выпадая в тканях, амилоид вытесняет специализированные клетки паренхиматозных органов и замещает клетки стенки сосудов. Его рост напоминает рост опухоли. Генерализованный амилоидоз может быть проявлением самостоятельного заболевания — приобретенного (первичный, или идиопатический; старческий) или наследственного (наследственный), может быть осложнением болезни или второй болезнью (вторичный, или реактивный, амилоидоз). В последние два десятилетия благодаря успехам биохимии, иммунологии, генетики и молекулярной биологии в проблеме амилоидоза получены новые факты, позволяющие пересмотреть многие ее положения. 1. Доказана гетерогенность белка амилоидных фибрилл — выделено несколько видов специфического фибриллярного белка амилоида — АА, AL, FAP, ASC1. Это значит, что единого амилоида при генерализованном амилоидозе нет, существу амилоидоза — АА-, AL-, FAP-, ASC1-амилоидоз. Выделение таких групп амилоидоза оказалось весьма перспективным. Оно показало, что каждая из этих групп представлена этиологически разными его формами (схема 5). Схема 5. Соотношение этиологических и био(гисто)химических форм генерализованного амилоидоза ФОРМЫ АМИЛОИДОЗА Био(гисто)химические Этиологические АА-амилоидоз Вторичный (реактивный) Наследственный (периодическая болезнь, синдром Макла — Уэльса) AL-амилоидоз Первичный (идиопатический) Вторичный (моноклоново-белковый, при неопластической плазмоклеточной дискразии) FAP-амилоидоз Наследственный (семейная амилоидная полинейропатия) ASC1- амилоидоз Старческий системный (генерализованный) АА-амилоидоз включает не только вторичные (реактивные) формы, но и наследственные — периодическую болезнь и синдром Макла — Уэльса. AL-амилоидоз представлен первичным амилоидозом и амилоидозом при так называемой неопластической плазмоклеточной дискразии, или "моноклоново-белковым амилоидозом". FAP-амилоидоз — это семейная амилоидная полинейропатия, этническая принадлежность которой оказалась достаточно широкой: заболевание встречается не только в Португалии, как считали раньше, но и в Японии, Швеции, Англии, Польше, Греции, Германии, Израиле. ASC1-амилоидоз — старческий системный амилоидоз. Эти данные свидетельствуют об условности деления амилоидоза в зависимости от возможной причины его возникновения, т.е. на первичный, вторичный, наследственный, старческий и т.д. Условность такого деления подтверждается и рядом более убедительных фактов. Так, среди больных первичным амилоидозом, с одной стороны, найдена подгруппа с плазмоклеточной дискразией, с другой — случаи семейного амилоидоза; среди форм АА-амилоидоза, помимо вторичного и наследственного, описываются идиопатические случаи. 2. Идентифицированы циркулирующие в крови предшественники белка фибрилл амилоида при генерализованных формах амилоидоза АА-, AL-,FAP-, ASC1-формы. Предшественником АА-белка амилоидных фибрилл является а-глобулин, названный сывороточным амилоидным белком — SАА. Доказана возможность трансформации его в АА-белок с образованием амилоидных фибрилл in vivo. У человека и животных SAA ведет себя подобно "острофазному" белку: содержание его в сыворотке после применения воспалительного стимула повышается в несколько сотен раз. Установлено влияние SAA на активность киллеров, взаимодействие Т-лимфоцитов с макрофагами, синтез антител плазматическими клетками. Таким образом, можно говорить об определенной связи между содержанием SAA в сыворотке крови и состоянием систем моноцитарного фагоцита и лимфоцита. Это открывает перспективы изучения патогенеза АА-амилоидоза. SAA синтезируется главным образом гепатоцитами, хотя показана возможность синтеза его и другими клетками — фибробластами и полиморфно-ядерными лейкоцитами. В гепатоцитах осуществляются не только синтез, но и деградация SAA до его субъединиц, что определяет постоянство содержания SAA в сыворотке крови в ничтожных (до 1 мкг/мл) количествах в нормальных условиях. Содержание SAA в сыворотке крови зависит и от активности макрофагальной системы. Часть циркулирующего SAA фильтруется в почечных клубочках и реабсорбируется — это второй путь метаболизма SAA. Об этом свидетельствует обнаружение SAA в подоцитах и мезангиальных клетках, способных к фагоцитозу. Второй путь метаболизма SAA позволяет понять особую "заинтересованность" почек при АА-амилоидозе ("нефропатический амилоидоз"). Предшественником AL-белка амилоидных фибрилл (моноклонального белка) являются λ- и κ-легкие цепи иммуноглобулинов или их фрагменты. Среди легких цепей найдены определенные, более амилоидогенные типы. Белок амилоидных фибрилл образуется из сывороточного предшественника либо при нарушении деградации моноклональных легких цепей, что ведет к появлению промежуточных полипептидов, способных к агрегации в фибриллы, либо при возникновении легких цепей с особыми структурами в связи с "аминокислотными заменами". Один из этих механизмов предусматривает участие в построении амилоидных фибрилл клеток, способных к деградации, другой — способных к синтезу белка. При FAP-амилоидозе белок фибрилл амилоида образуется из преалъбумина плазмы с различными "аминокислотными заменами" при разных этнических вариантах FAP, что пытаются объяснить образованием мутантов. Выявленное высокое содержание преальбумина в цереброспинальной (спинномозговой) жидкости связывают с активным синтезом его хориоидальным сплетением. Этим объясняют особенности клинической симптоматики полинейропатии. В сыворотке крови у больных FAP-амилоидозом и в группе риска уровень преальбумина снижен в 4—6 раз, что отличает этот амилоидоз от других форм генерализованного амилоидоза и свидетельствует о расходовании предшественника на построение белка амилоидных фибрилл. Однако депрессию преальбумина при FAP-амилоидозе можно рассматривать и как генетически запрограммированное нарушение его метаболизма. При старческом системном амилоидозе предшественником фибриллярного белка ASC1, который рассматривается как мутантный белок, также является сывороточный пре-альбумин. Считают, что в связи с нарушением метаболизма преальбумина в пожилом и старческом возрасте склонность к образованию ASC1 из циркулирующего в крови предшественника повышается, чем и объясняют снижение содержания преальбумина в сыворотке при этой форме амилоидоза. Не исключается аффинность предшественника к определенным тканям (кардиомиоцит, гладкая мышечная клетка сосудов). На основании приведенных данных можно обосновать патогенез АА- и AL-форм генерализованного амилоидоза. Патогенез АА-амилоидоза. Основные этапы патогенеза АА-амилоидоза следующие (схема 6): ▲ стимуляция синтеза SAA (печень) интерлейкином-1 в результате активации системы моноцитарных фагоцитов, что ведет к резкому увеличению содержания предшественника АА-белка в плазме крови; ▲ усиленная, но неполная ферментативная дегратация SAA макрофагами, появление белка АА; ▲ сборка на поверхности макрофагов-амилоидобластов фибрилл амилоида из фрагментов деградирующего SAA (белка АА) под воздействием амилоидстимулирующего фактора (АСФ) и при участии деградирующей активности сыворотки (ДАС). В этой системе следует найти место второму (почечному) пути метаболизма SAA, о котором уже упоминалось и которой делает понятным преимущественное поражение почек при АА-амилоидозе. Схема 6. Патогенез АА-амилоидоза 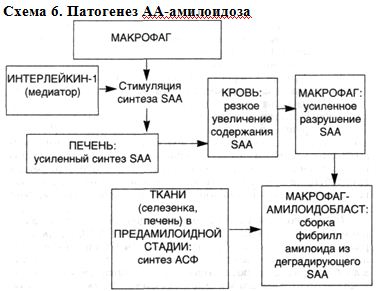 Патогенез AL-амилоидоза. Основными звеньями патогенеза AL-амилоидоза (схема 7) можно считать следующие: ▲ синтез легких цепей иммуноглобулинов как проявление "плазмоклеточной дискразии"; ▲ образование амилоидных фибрилл из легких цепей плазматическими, миеломными (опухолевыми) клетками и макрофагами в условиях нарушенной деградации моноклональных легких цепей или появления легких цепей с особыми структурами ("аминокислотные замены"). При этом между клетками-амилоидобластами — миеломными (плазматическими) и макрофагами — возникает подчиненное механизму амилоидогенеза взаимодействие. Схема 7. Патогенез AL-амилоидоза 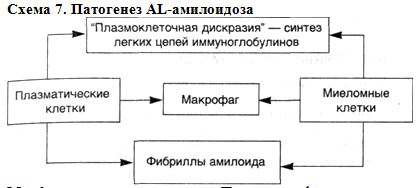 Морфологические изменения. При разных формах генерализованного амилоидоза они различны. При АА-амилоидозе (вторичный амилоидоз, при периодической болезни и синдроме Макла — Уэльса) поражаются преимущественно паренхиматозные органы ("паренхиматозный амилоидоз") — селезенка, почки, печень, надпочечники, кишечник. Они увеличиваются в размерах, становятся очень плотными, приобретают сальный вид — "сальная печень", "сальная селезенка" и т.д. Для AL-амилоидоза (первичный и моноклоново-белковый при хронических парапротеинемических лимфолейкозах) и ASC1-амилоидоза (генерализованный старческий амилоидоз) характерно поражение преимущественно мезодермалъных тканей ("мезенхимальный амилоидоз") — сердца и сосудов (особенно коронарных, легочных и кишечных), поперечнополосатых и гладких мышц, нервов и кожи. Амилоидная кардиомегалия — основной морфологический признак, который при AL-амилоидозе выражен значительно сильнее, чем при ASC1. При ASC1-амилоидозе возможно сочетание поражения сердца, мозга и панкреатических островков, островков поджелудочной железы. FAP-амилоидоз — наследственная амилоидная нейропатия с поражением нервов рук и ног — не исчерпывает все формы наследственного амилоидоза. Выделяют также нефропатический (периодическая болезнь, синдром Макла — Уэльса) и кардиопатический (подобный первичному) наследственный амилоидоз. Таким образом, можно утверждать, что существует гетерогенность как био(гисто)химических свойств фибриллярного белка амилоида, так и патогенеза генерализованного амилоидоза. Это исходно обусловлено различными причинами, т.е. этиологией амилоидоза. С гетерогенностью генерализованного амилоидоза связано огромное разнообразие его клинико-морфологических проявлений в виде самостоятельных заболеваний и осложнений многих других болезней, которые выступают нередко в качестве вторых болезней. Все это позволяет говорить не об амилоидозе, а об амилоидозах, которые, являясь системными (генерализованными), становятся морфологической и патогенетической основой системной дезорганизации соединительной ткани, в основе которой лежит прогрессирующий синтез аномального фибриллярного белка. |
| Вы можете прокомментировать статью | |
