| Академия » Статьи » Лекции » Анатомия |
ПАРЕНХИМАТОЗНЫЕ ДИСТРОФИИ: ДИСПРОТЕИНОЗЫ И ЛИПИДОЗЫ
|
ПАРЕНХИМАТОЗНЫЕ ДИСТРОФИИ: ДИСПРОТЕИНОЗЫ И ЛИПИДОЗЫ • Дистрофия (греч. dys — нарушение и trophe — питание) — патологический процесс, в основе которого лежат нарушения тканевого (клеточного) обмена, ведущие к структурным изменениям. Поэтому дистрофия рассматривается как один из видов повреждения. Непосредственной причиной развития дистрофий являются нарушения клеточных и внеклеточных механизмов трофики. Среди них выделяют: ▲ расстройства ауторегуляции клетки, ведущие к энергетическому ее дефициту и нарушению ферментативных процессов в клетке; в таких случаях ферментопатия (приобретенная или наследственная) становится основным патогенетическим звеном и выражением дистрофии; а ч нарушения функции транспортных систем трофики (кровь, лимфа, микроциркуляторное русло, интерстициальная ткань), обусловливающие развитие гипоксии, которая становится ведущей в патогенезе дисциркуляторных дистрофий; а расстройства интегративных систем трофики, т.е. эндокринной и нервной ее регуляции, что определяет развитие эндокринных и церебральных дистрофий. Среди морфогенетических механизмов дистрофии различают инфильтрацию, декомпозицию, извращенный синтез, трансформацию. Инфильтрация — избыточное проникновение продуктов обмена из крови и лимфы в клетки или межклеточное вещество; последующее накопление их обусловлено недостаточностью ферментных систем, метаболизирующих эти продукты. Декомпозиция (фанероз) — распад ультраструктур клеток и межклеточного вещества, ведущий к нарушению тканевого (клеточного) метаболизма и накоплению продуктов нарушенного обмена в ткани (клетке). Извращенный синтез — синтез в ткани (клетке) веществ, не встречающихся в них в норме. Трансформация — образование продуктов одного вида обмена из общих исходных продуктов, которые идут на построение белков, жиров и углеводов. Инфильтрация и декомпозиция нередко являются последовательными стадиями морфогенеза той или иной дистрофии. В связи со структурно-функциональными особенностями в некоторых органах и тканях преобладает какой-либо один из морфогенетических механизмов, что позволяет говорить об ортологии (от греч. orthos — типичный) дистрофий. Классификация дистрофий. Различают следующие виды дистрофий: ▲ Паренхиматозные, стромально-сосудистые и смешанные дистрофии — в зависимости от преобладания морфологических изменений в специализированных элементах паренхимы или строме и сосудах; ▲ Белковые (диспротеинозы), жировые (липидозы), углеводные и минеральные дистрофии — в зависимости от преобладания того или иного вида обмена; ▲ Общие (системные) и местные — в зависимости от распространенности процесса; ▲ Приобретенные и наследственные. При паренхиматозных дистрофиях возникают нарушения обмена высокоспециализированных в отношении функции клеток (клеточных коопераций) паренхиматозных органов. В разных органах (почки, печень, сердце) при развитии одного и того же вида дистрофии участвуют различные механизмы. При этом развитие того или иного вида паренхиматозной дистрофии обусловлено не только своеобразием повреждающего фактора, но и структурно-функциональными особенностями специализированных клеток (клеточных коопераций) паренхиматозных органов. Переход одного вида паренхиматозной дистрофии в другой исключается, возможно лишь сочетание разных видов этой дистрофии. В основе развития паренхиматозных дистрофий лежат нарушения клеточных механизмов трофики, т.е. ферментопатия, которая может быть приобретенной или наследственной. Наследственная ферментопатия лежит в основе большой группы болезней накопления, или тезаурисмозов. Различные виды паренхиматозных дистрофий составляют морфологическую сущность недостаточности определенного механизма, служащего выполнению клеткой (нефроцит, гепатоцит, кардиомиоцит) специализированной функции. Поэтому паренхиматозная дистрофия, чаще белковая или жировая, лежит в основе клинического синдрома, отражающего функциональную недостаточность паренхиматозного органа (почки, печень, сердце). ПАРЕНХИМАТОЗНЫЕ ДИСПРОТЕИНОЗЫ • Паренхиматозные диспротеинозы, или паренхиматозные белковые дистрофии, характеризуются нарушением обмена цитоплазматических белков, которые находятся в свободном или связанном состоянии. Связанные белки входят в состав липопротеидных комплексов мембран клетки; к свободным белкам относятся главным образом ферменты. Развитие паренхиматозных диспротеинозов сопровождается изменением физико-химического состояния белков, появлением в цитоплазме включений белковой природы. Нарушение обмена белков нередко сочетается с расстройством водно-электролитного обмена в цитоплазме, изменением коллоидно-осмотического давления, что ведет к ее гидратации. Паренхиматозные диспротеинозы морфологически представлены гиалиново-капельной и гидропической дистрофией. Исходом каждой из них может быть некроз клетки: коагуляционный фокальный или тотальный — при гиалиново-капельной и колликвационный фокальный (баллонная дистрофия) или тотальный — при гидропической дистрофии. Для паренхиматозных диспротеинозов характерны лишь микроскопически обнаруживаемые изменения — накопление в цитоплазме гиалиновых капель при гиалиново-капельной дистрофии и увеличение обмена клеток за счет гидратации цитоплазмы и появления вакуолей, содержащих прозрачную жидкость, при гидропической дистрофии. Механизм развития того или иного вида паренхиматозного диспротеиноза может быть раскрыт только на основании анализа особенностей функционирования систем клетки (клеточных коопераций), обеспечивающих специализированную функцию органа [Серов В.В., 1972]. Это может быть показано при анализе паренхиматозных диспротеинозов почек и печени. Почки. Развитие гиалиново-капельной и гидропической дистрофии нефроцитов при нефротическом синдроме (сочетание массивной протеинурии с отеками, гипо- и диспротеинемией, гиперхолестеринемией и гиперлипопротеидемией) связывают с механизмом инфильтрации, но этого недостаточно, чтобы понять суть этих дистрофий. Такая возможность возникает лишь при морфологическом анализе состояния при нефротическом синдроме мембранно-ферментных систем нефроцита, ответственного за реабсорбцию белка и воды. Рис. 1. Вакуолярно-лизосомальный аппарат реабсорбции белка нефроцитом. 1 — вакуоли, заполненные белком; 2 — предсуществующие лизосомы; 3 — слияние лизосомы с вакуолью, заполненной белком; 4 — лизосома с инкорпорируемым и перевариваемым белком; ПК — пластинчатый комплекс. Стрелками показаны последовательные этапы реабсорбции белка. 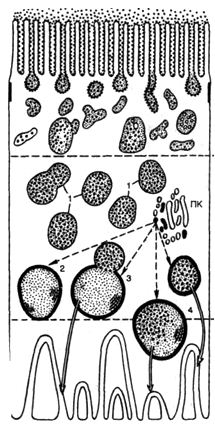 Как известно, проксимальная реабсорбция белка осуществляется с помощью вакуолярно-лизосомальной системы нефроцитов — системы переваривания фильтрующегося в норме белка и его утилизации (рис. 1). Реабсорбция воды и натрия связана с ба-зальным лабиринтом и обеспечивается главным образом натрий-и калийзависимыми АТФазами. Выявлено, что при гиалиново-капельной дистрофии нефроцитов накопление белковых включений в цитоплазме и ее деструкция обусловлены несостоятельностью вакуолярнолизосомального аппарата реабсорбции белка в условиях повышенной порозности гломерулярного фильтра при нефротическом синдроме. Сами гиалиновые включения представляют собой заполненные белками, "задыхающиеся" и распадающиеся лизосомы, что определяет высвобождение их ферментов и вторичную деструкцию. Гидропическая дистрофия нефроцитов обусловлена недостаточностью другой системы реабсорбции — системы базального лабиринта, который в тех же условиях повышенной порозности гломерулярной базальной мембраны переполняется поступающей в клетку водой и "поднимается" к щеточной каемке, разрушая мембраны и образуя баллонные структуры — гидропическая дистрофия становится баллонной (фокальный колликвационный некроз). Как видно, механизмы развития гиалиново-капельной и гидропической дистрофии в почках при нефротическом синдроме разные, коль скоро они отражают недостаточность разных систем проксимальной реабсорбции. Печень. При оценке гидропической дистрофии гепатоцитов следует руководствоваться тем же морфологическим анализом особенностей функционирования печеночных клеток, обеспечивающих специализированные функции органа. Известно, что для выполнения синтетической (белковосинтетической) и антитоксической функции гепатоциты детерминированы структурно: темные гепатоциты периферии долек богаты ультраструктурами синтеза, светлые гепатоциты центров долек — ультраструктурами детоксикации и гидролиза. При воздействии на печень вируса гепатита В избирательно реагируют темные, а токсичных веществ — светлые гепатоциты. При этом дистрофия их отражает разные по своей сути процессы: гидропическая дистрофия темных гепатоцитов — извращенную белковосинтетическую функцию, подчиненную репродукции вируса, та же гидропическая дистрофия светлых гепатоцитов — недостаточность системы детоксикации. На основании этих данных возникает вопрос о значении функциональной гетерогенности структуры при морфофункциональной оценке дистрофического процесса. При паренхиматозных диспротеинозах в гепатоцитах могут появляться гиалиноподобные включения — развивается процесс, близкий к гиалиново-капельной дистрофии. Среди этих включений наибольший интерес представляет алкогольный гиалин (тельца Маллори). Его находят в гепатоцитах чаще при остром алкогольном гепатите, а также при первичном билиарном циррозе печени, гепатоме, холестазе. Эти тельца располагаются обычно перинуклеарно в виде ацидофильных глыбок или сетчатых масс. Электронная микроскопия подтверждает фибриллярное строение этого белка, который является продуктом синтеза гепатоцитов. Алкогольный гиалин определяет ряд реакций как в печени, так и за ее пределами, что обусловлено рядом его свойств. Он обладает хемотаксическими свойствами и определяет прежде всего лейкотаксис, поэтому он окружен, как правило, полиморфно-ядерными лейкоцитами (характерный признак острого алкогольного гепатита). Алкогольный гиалин оказывает цитолитическое действие на гепатоциты, с чем связано развитие в печени своеобразного склерозирующего гиалинового некроза", и коллагеностимулирующее действие, определяя хроническое прогрессирующее течение алкогольного гепатита и развитие цирроза печени. Иммуногенные свойства алкогольного гиалина позволяют ему участвовать в гуморальных и клеточных иммунных реакциях, что и обусловливает развитие системной патологии. Антиген алкогольного гиалина обнаруживается в составе циркулирующих иммунных комплексов, с чем связано развитие иммунокомплексного поражения почек — гломерулонефрита. Иммунокомплексное повреждение почек, как видно, связано с синтезом алкогольного гиалина печенью, циркуляцией антигена алкогольного гиалина в крови с последующей фиксацией его в составе иммунных комплексов в клубочках почек. ■ Таким образом, можно говорить о системных проявлениях паренхиматозных диспротеинозов, в частности своеобразной гиалиново-капельной дистрофии гепатоцитов (алкогольный гиалин). Наследственные паренхиматозные диспротеинозы Наследственные паренхиматозные диспротеинозы обусловлены нарушением внутриклеточного метаболизма аминокислот и представлены цистинозом, тирозинозом и фенилпировиноградной олигофренией (фенилкетонурией). Поражаются печень, почки, селезенка, костный мозг и центральная нервная система. ПАРЕНХИМАТОЗНЫЕ ЛИПИДОЗЫ Паренхиматозные липидозы, или паренхиматозные жировые дистрофии, характеризующиеся нарушением обмена жиров в цитоплазме, морфологически проявляются увеличением их количества в клетках, где они встречаются в нормальных условиях, появлением их там, где они обычно не встречаются, и образованием жиров необычного химического состава. Чаще в клетках накапливаются нейтральные жиры. Термином "липиды", как известно, обозначают все жиры, включая сложные лабильные жиробелковые комплексы — липоиды, составляющие основу мембранных структур клетки. Помимо липоидов, к липидам относят и нейтральные жиры, являющиеся сложными эфирами жирных кислот и глицерина. Преобладание морфогенетического механизм а при паренхиматозном липидозе зависит от причины, вызвавшей дистрофию, и структурно-функциональных особенностей органа. Однако в большинстве случаев бывает трудно выделить главный из них, так как отмечаются смена одного морфогенетического механизма другим либо их сочетания., Паренхиматозная жировая дистрофия наиболее часто встречается в печени, миокарде и почках. Печень. О жировой дистрофии печени, которая по сравнению с другими липидозами паренхиматозных органов встречается особенно часто, говорят в тех случаях, когда жир, преимущественно нейтральный, содержит более 50 % гепатоцитов. Выделяют три стадии жировой печени [Kalk H., 1965]: "чистая" жировая печень, жировая печень с мезенхимальной реакцией, фиброз и цирроз печени. Непосредственной причиной накопления нейтральных жиров в печени является дезорганизация ферментативных процессов на том или ином этапе обмена липидов, которая проявляется в следующих ситуациях: ▲ при чрезмерном поступлении в клетку жирных кислот или повышенном их синтезе в гепатоците, что создает относительный дефицит ферментов; ▲ при воздействии на клетку токсичных веществ, блокирующих окисление жирных кислот, синтез апопротеинов; ▲ при недостаточном поступлении в гепатоциты аминокислот, необходимых для синтеза фосфолипидов и липопротеидов (схема 3). 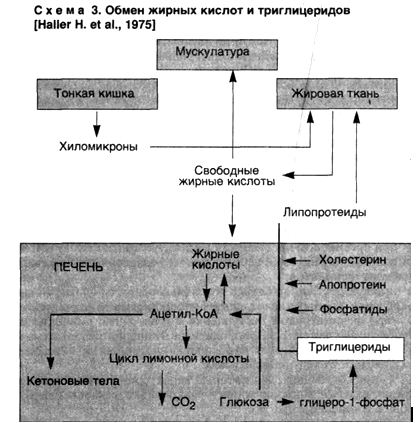 Из сказанного следует, что жировая дистрофия печени может развиваться в следующих случаях: 1) при состояниях, для которых характерен высокий уровень жирных кислот в плазме крови — алкоголизм, сахарный диабет, общее ожирение и др.; 2) при воздействии на гепатоциты токсичных веществ — этанола, четыреххлористого углерода, фосфора и др.; 3) при нарушении питания вследствие недостатка белка в пище (алипотропное ожирение печени) или заболеваний желудочно-кишечного тракта; 4) при генетических дефектах ферментов, участвующих в жировом обмене — наследственные липидозы. Более подробного рассмотрения заслуживает жировая дистрофия печени при алкоголизме, сахарном диабете и интоксикациях. Алкоголизм. Установлено, что среди других причин развития жировой печени этанолу отводится от 30 до 50 %. Этанол усиливает мобилизацию жира из депо, увеличивает синтез жирных кислот в гепатоцитах, усиливает этерификацию жирных кислот до триглицеридов, снижает уровень окисления жирных кислот, уменьшает синтез и освобождение липопротеидов, а также проницаемость клеточной мембраны гепатоцита в связи с усилением синтеза и накоплением холестерина. Отложения жира в алкогольной печени могут быть очаговыми и диффузными. В случае диффузного ожирения алкогольная печень увеличена в размерах, дряблая, охряно-желтая. При гистологическом исследовании в зависимости от размеров жировых капель различают мелкокапельную, среднекапельную и крупнокапельную дистрофию гепатоцитов. При крупнокапельной жировой дистрофии — крайнем выражении дистрофии — ядро гепатоцита оттесняется к наружной мембране клетки. Ободок цитоплазмы, свободной от жировых включений, остается структурно и функционально сохранным: при электронной микроскопии органеллы его изменены мало, в нем высоки содержание гликогена, РНК и активность ферментов гликолиза, пентозного шунта, дезаминирования; достаточна и активность сукцинатдегидрогеназы. Авторадиография подтверждает сохранение синтетических процессов в ожиревших гепатоцитах. Видимо, возможный исход алкогольной жировой печени (алкогольного стеатоза печени) объясняется тем, прекратит ли больной употребление алкоголя или будет продолжать злоупотреблять им. На основании результатов исследования повторных биоптатов печени у больных алкоголизмом показано, что при полной абстиненции жир исчезает из печени через 2—4 нед, а прогрессирование алкогольного стеатоза ведет к формированию цирроза печени, при этом большое значение имеют повторные атаки острого алкогольного гепатита. Сахарный диабет. Жировая дистрофия печени у больных сахарным диабетом встречается в 50—75 % случаев, причем выраженность стеатоза коррелирует с возрастом (при юношеской форме сахарного диабета жировая печень встречается редко), массой тела больных и тяжестью кетоацидоза. Развитие стеатоза печени при сахарном диабете обусловлено усиленными мобилизацией жира из жировых депо, транспортом их в печень, нарушением синтеза фосфолипидов и окисления жирных кислот. Усиленный липолиз обусловлен недостатком инсулина, который является антилиполитическим гормоном, — липолитические гормоны "берут верх" над антилиполитическими. В результате липолиза в крови увеличивается содержание жирных кислот, а в печени усиливается синтез липопротеидов. Но печень вследствие недостаточного синтеза апопротеина не в состоянии полностью усвоить поступающие жирные кислоты, идущие на построение липопротеидов. Избыток жирных кислот ресинтезируется в печени в триглицериды. При сахарном диабете, сопровождающемся общим ожирением (что отмечается довольно часто), стеатоз печени усиливается в связи с избыточным поступлением жиров и углеводов с пищей. При этом основным механизмом поступления жира в печень остается липолиз, ведущий к гиперлипидемии. Морфологической особенностью жировой дистрофии печени при сахарном диабете является вакуолизация ядер ожиревших гепатоцитов за счет накопления в них гликогена — "дырчатые", или "гликогенные", ядра. Жир в цитоплазме гепатоцитов (стеатоз) и "дырчатые" их ядра (накопление гликогена) — характерные гистологические признаки диабетической жировой дистрофии печени. Интоксикации. Жировая дистрофия печени развивается при воздействии на организм таких токсичных веществ, как четыреххлористый углерод, гидразин-сульфат, тетрахлорэтан, тринитротомзол, ДДТ, фосфор, а также ряда лекарственных средств (тетрациклины, стероиды, барбитураты, метотрексат и др.). В этих условиях накопление липидов в гепатоцитах обусловлено, как правило, нарушением синтеза белка (апопротеина) вследствие блокады их ферментных систем. Недостаток апопротеина вызывает нарушение синтеза липопротеидов, способных проникать через наружную мембрану гепатоцитов. Задержка липидов в цитозоле приводит к образованию триглицеридов. Накопление жира в гепатоцитах связано и с распадом липопротеидных комплексов мембран гепатоцитов, т.е. с механизмом фанероза. Миокард. Развитие жировой дистрофии миокарда связывают с тремя основными механизмами: ▲ повышенным поступлением жирных кислот в кардиомиоциты; ▲ нарушением обмена жиров в этих клетках; ▲ распадом липопротеидных комплексов внутриклеточных структур, т.е. фанерозом. Основой этих трех механизмов жировой Дистрофии кардиомиоцитов является энергетический дефицит миокарда. Известно, что в кардиомиоциты липиды поступают в виде жирных кислот, освобождающихся с помощью липопротеидлипазы от плазменных триглицеридов или связей с альбуминами. Жирные кислоты используются миокардом для энергетических нужд (что достигается их β-окислением в митохондриях) и для построения структурных фосфолипидов. Из этого следует, что при любых состояниях, сопровождающихся энергетическим дефицитом, усиливается поступление в миокард жирных кислот, из которых синтезируются нейтральные жиры. Механизм фанероза в развитии жировой дистрофии кардиомиоцитов заключается не в высвобождении липидов из липопротеидных комплексов мембранных структур клетки, а в нарушении окисления поступающих в избытке в клетку жирных кислот при деструкции ее митохондрий. Причины развития жировой дистрофии миокарда следующие: 1) гипоксия (при анемиях, хронической сердечно-сосудистой недостаточности); 2) интоксикации (дифтерийная, алкогольная, отравление фосфором, мышьяком, хлороформом и др.). Гипоксия. Это наиболее частая причина жировой дистрофии миокарда, поскольку гипоксия ведет к энергетическому дефициту высокоспециализированных тканей, к которым относится миокард. Недостаток кислорода нарушает процессы окислительного фосфорилирования в кардиомиоцитах, что приводит к переключению обмена миокарда на анаэробный гликолиз и резкому снижению количества АТФ. Дефицит энергии усиливается в связи с нарастающим ацидозом ткани; развивается повреждение митохондрий, нарушается окисление жирных кислот, и липиды накапливаются в кардиомиоцитах чаще в виде мелких капель (различают также и пылевидное ожирение миокарда). Жировая дистрофия миокарда чаще имеет очаговый характер — содержащие жир кардиомиоциты расположены преимущественно по ходу венозного колена капилляров и мелких вен, где гипоксический фактор наиболее резко выражен. Очаговостью поражения объясняется своеобразный внешний вид сердца: со стороны эндокарда, особенно в области сосочковых мышц, видна желтовато-белая исчерченность ("тигровое сердце"); миокард дряблый, бледно-желтый, камеры сердца растянуты, размеры его Несколько увеличены. Интоксикации. Наиболее изучена жировая дистрофия миокарда при дифтерийной и алкогольной интоксикации. При дифтерийной интоксикации накопление липидов в кардиомиоцитах обусловлено снижением интенсивности их окисления вследствие как недостатка карнитина, так и повреждения митохондрий (рис. 2). При алкогольной интоксикации также имеют место снижение интенсивности окисления жирных кислот в кардиомиоцитах и деструкция их митохондрий, что ведет к резкому уменьшению активности ферментов. Рис. 2. Транспорт жирных кислот через митохондриальную мембрану (схема). Морфологические изменения сердца при интоксикации подобны таковым при гипоксии, но при дифтерии по сравнению с алкоголизмом они выражены сильнее. Почки. Следует помнить, что нейтральные жиры обнаруживаются в эпителии узкого сегмента и собирательных трубочек и в физиологических условиях. О жировой дистрофии почек говорят в тех случаях, когда липиды (нейтральные жиры, холестерин, фосфолипиды) появляются в эпителии канальцев главных отделов нефрона — проксимальных и дистальных. Наиболее часто жировая дистрофия почек встречается при нефротическом синдроме и хронической почечной недостаточности, реже — при инфекциях и интоксикациях. Нефротический синдром. Как уже упоминалось ранее, нефротический синдром характеризуется не только массивной протеинурией, обусловливающей развитие отеков и гипо-, диспротеинемии, но и гиперлипидемией, повышением в крови Уровня триглицеридов, холестерина и фосфолипидов. Гиперлипидемию в этих случаях объясняют увеличением синтеза холестерина и мобилизацией жира из жировых депо/снижением активности липопротеидлипазы и холестеринлецитинацетилтрансферазы в сыворотке крови, усилением синтеза липидов в почках вследствие угнетения почечной липолитическои активности. Понятно, что гиперлипидемия обусловливает липидурию, главным образом за счет липопротеидов. В условиях характерной для нефротического синдрома повышенной проницаемости гломерулярного фильтра липиды подвергаются повышенной резорбции эпителием канальцев, загружая не только цитоплазму нефроцитов, но и строму почки. Жировая дистрофия нефроцитов при нефротическом синдроме присоединяется к гиалиново-капельной и гидропической, о которых уже шла речь ранее. Хроническая почечная недостаточность. При этом синдроме уровень триглицеридов и холестерина в крови также повышен. Это связывают со снижением активности липопротеидлипазы и уменьшением утилизации/глюкозы, что приводит к усилению липолиза. Снижение утилизации глюкозы обусловлено дефицитом белка в пищевом рационе больных с хронической почечной недостаточностью (уремией). Дефицит белка подавляет синтез ферментов, необходимых для процессов окисления. Морфологические изменения почек при жировой их дистрофии достаточно характерны. При микроскопическом исследовании липиды видны в цитоплазме эпителия канальцев и строме почки в виде капель (нейтральный жир) или двояко-преломляющих кристаллов (холестерин); Почки при нефротическом синдроме увеличены, дряблые, с желтым крапом на поверхности (при амилоидозе почек, сопровождающемся нефротическим синдромом, они плотные, с сальном блеском на разрезе). При хронической почечной недостаточности почки уменьшены, чаще с зернистой поверхностью, серо-желтые, с истонченным корковым веществом. Наследственные паренхиматозные липидозы Наследственные паренхиматозные липидозы, или системные липидозы, возникают вследствие наследственного дефицита ферментов, участвующих в метаболизме определенных липидов (наследственные ферментопатии). Поскольку дефицит фермента определяет накопление метаболизируемого им субстрата, системные липидозы относят к тезаурисмозам, или болезням накопления. Среди системных липидозов различают цереброзидлипидоз (болезнь Гоше), сфингомиелинлипидоз (болезнь Нимана — Пика),ганглиозидлипидоз (болезнь Тея — Сакса), генерализованный ганглиозидоз (болезнь Нормана — Ландинга) и др. Чаще всего страдают» печень, селезенка, костный мозг и центральная нервная система. Морфологическому диагнозу помогают обнаруживаемые в тканях характерные для того или иного вида липидоза клетки (клетки Гоше, клетки Пика). |
| Вы можете прокомментировать статью | |
